
Multiple endocrine neoplasia type 1 (MEN-1) is a rare inherited autosomal dominant disease which manifests itself with at least one clinical scenario before 45 years of age. The value of somatostatin analogue therapy is unknown in the treatment of non-functioning pancreatic tumours and a few studies have been published in this field. We report a young patient with MEN-1 with multiple gastric and pancreatic neuroendocrine tumors that was treated with monthly injection of Sandostatin LAR before and After Distal Pancreatectomy and partial gastrectomy.
Introduction:
Multiple endocrine neoplasia type 1 (MEN-1) is a rare inherited autosomal dominant disease which manifests itself with at least one clinical scenario before 45 years of age. Patients with MEN-1 develop different combinations of functional and non-functional tumors of parathyroid glands (90–97%), pancreatic islet cells and duodenum (30–80%), and the anterior pituitary gland (20–65%). Gastrointestinal neuroendocrine tumors (GI-NETs) are rare neoplasms, like all NETs. Gastric NETs (G-NETs) and duodenal NETs (D-NETs) are the common types of upper GI-NETs based on tumor location. Type 2 G-NETs make 5%-6% of all G-NETs and they are associated with MEN-1 and Zollinger-Ellison syndrome (MEN1-ZES).
The value of somatostatin analogue therapy is unknown in the treatment of non-functioning pancreatic tumours and a few studies have been published in this field.
We report a young patient with MEN-1 with multiple gastric and pancreatic neuroendocrine tumors that was treated with monthly injection of Sandostatin LAR before and After Distal Pancreatectomy and partial gastrectomy.
Case presentation
A 31‑year‑old lady was referred to our clinic, with intermittent abdominal pain and weight loss that had been present for 2 months. The symptoms could be temporarily relieved by symptomatic treatment. She had no complaints of lethargy, depression, confusion, anorexia, constipation, diarrhea, nausea; vomiting, oligomenorrhea, amenorrhea, galactorrhea, visual disturbances, sexual dysfunction. Patient’s medical and family histories were unremarkable.
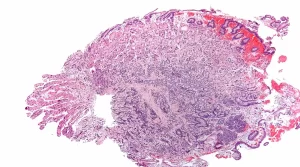
An upper gastrointestinal (UGI) endoscopy revealed mucosal congestion, diffuse erythema and nodularity in stomach and in pathology showed well differentiated neuroendocrine tumours. Subsequent endoscopy combined with Endoscopic ultrasound (EUS) (with fine needle aspiration, FNA) showed multiple small lesions at gastric wall and a small hypoechoic lesion in pancreatic body. Pathology reports of her stomach showed well differentiated neuroendocrine tumours with Ki-67 < 2% and the EUS-FNA cytology of pancreatic lesion was suggestive for neuroendocrine tumor and negative for any histological evidence of malignancy. On immunohistochemical (IHC) the tumor was positive for synaptophysin, chromogranin A and Ki‑67 (2%).
Her 24-hour urinary 5-hydroxyindolacetic acid (5-HIAA) was in the normal range, serum chromogranin A: 2942 ug/l (Nl: 19-98ug/l),serum gastrin: 600 pg/mL (normal <100 pg/mL), serum calcium: 12.9 mg/ dl (Nl: 8.6-10.3mg/dl), serum phosphate: 2.6 mg/dl ( 2.5-4.5) and parathormone (PTH): 300 pg/ml (Nl: 15-65pg/ml), T 4:7.9ug/dl (Nl: 5.1-14.1 ug/dl), TSH: 0.6 MIu/l (Nl: 0.3-3.6 MIu/l), FSH: 2.3 MIu/ml, LH:6.3 MIu/ml, prolactin: 20 ng/ml (Nl: 6-30ng/ml), IGF1:196ng/ml (Nl: 108-247ng/ml).
Additional diagnostic evaluation for MEN-1 syndrome included parathyroid scintigraphy and magnetic resonance imaging (MRI) of the sella turcica were performed. Parathyroid scintigraphy (SPECT/CT) by TC99m MIBI was suggestive of double parathyroid adenoma below the lower poles of thyroid lobes which confirmed primary hyperparathyroidism and pituitary MRI showed abnormal signals in the right anterior gland measuring 4.1* 6.1 mm in favor of microadenoma.
Based on this evidence, the diagnosis of MEN-1 syndrome was confirmed. Three months later, total parathyroidectomy, thymectomy and parathyroid auto-
transplantation to the forearm was performed. Following surgery calcium level decreased to 7.9 mg/dl (8.6-10.3). Six months after surgery another EUS was performed which revealed numerous small hypoechoic polypoid lesions in the gastric body and antrum as well as six small mass lesions in pancreas. All of them were endosongraphically consistent with neuroendocrine tumours. She was started on Sandostatin LAR injections 30 mg every 4 weeks. After three months another EUS was done which revealed regression of the gastric lesions and stable tumours in the pancreas.
Sandostatin LAR was continued and none months later she underwent distal pancreatectomy with enucleation of tumors in the posterior head of pancreas, splenectomy and partial gastrectomy to decrease the gastric burden of the tumour.
A year after surgery, repeat endoscopy showed a few polyps less than 6 mm in the gastric corpus and antrum. On histopathologic examination, some of these polyps were reported as low grade neuroendocrine tumour and positive Ki-67 in 1 % of tumour cell nuclei. The patient was maintained on intramuscular Sandostatin LAR 20 mg monthly currently, after three years the patient remains well with no progression of the tumours
Discussion:
MEN1, a rare autosomal dominant disease, is reported in all age groups and more than 95% of patients show clinical symptoms by the fifth decade. The manifestations are mostly due to the secreted products of the tumours rather than local or metastatic effects. Non-familial (i.e. sporadic) form may occur in 8-14% of patients with MEN1. Parathyroid tumours are the first manifestation of MEN-1 in over 85% of patients. Anterior pituitary tumours that include prolactinoma, somatotropinomas, corticotropinomas, or nonfunctional adenomas happen in about 30% of patients. NETs, rare tumours of neuroendocrine cell origin, also occur in MEN-1. These multipotent cells able to produce and secrete active substances which can cause distinct clinical syndromes. Both functioning and nonfunctioning tumours can also produce symptoms due to their mass effects. These are usually slow growing tumours, but rapid with distant metastases may occur. In 50% of cases, the origin of these tumours is the GI tract or the pancreas (gastroenteropancreatic neuroendocrine neoplasms [GEP-NETs]). Gastrointestinal neuroendocrine tumours (GI-NETs) are rare neoplasms, but their incidence has been increasing in recent years. Gastric NETs (G-NETs) and duodenal NETs (D-NETs) are the common types of upper GI-NETs based on tumour location. G-NETs are classified into three subgroups: types 1, 2, and 3.
Type 1 G-NETs, are the most common subtype (70%-80% of all G-NETs), and associated with chronic atrophic gastritis, including autoimmune gastritis and Helicobacter pylori associated atrophic gastritis. Type 2 G-NETs make 5%-6% of all G-NETs and are associated with multiple endocrineneoplasia type 1 and Zollinger-Ellison syndrome (MEN1-ZES). Both type 1 and 2 G-NETs are related to hypergastrinemia, are small in size, occur in multiple numbers, and are usually benign. In contrast, type 3 G-NETs (10%-15%) are not associated with hypergastrinemia, are large-sized single tumours, and are usually malignant. Therefore, surgical resection and chemotherapy are generally necessary for type 3 G-NETs, while endoscopic resection and follow up, which are acceptable for the treatment of most type 1 and 2 G-NETs, are only acceptable for small and well differentiated type 3 G-NETs. Use of Endoscopic ultrasound (EUS) facilitates detection and localization of P-NETs as small as 1–2 mm. Considering their rarity, only small series or case reports have been published.
NETs often secrete vasoactive substances causing carcinoid syndrome. They also express somatostatin receptors. Octreotide and lanreotide are the two synthetic somatostatin analogs (SSA) used for both control of carcinoid symptoms and tumor progression in advanced inoperable disease. Octreotide acetate, developed in 1980s, has a shorter peptide length (of 8 aminoacids only) than somatostatin, a longer half-life (1.5 to 2 hours) and is more potent and resistant to degradation . Lanreotide, another SSA octapeptide initially developed in 1990s as a sustained release formulation has been reported to reduce symptoms by 50% when administered intramuscularly every 2 weeks. Both molecules have similar mechanisms of action with similar somatostatin receptors binding affinities and similar symptom control rates in carcinoid syndrome patients (55–75%). Although octreotide LAR (long acting octreotide) is FDA approved for carcinoid symptom control and lanreotide autogel is approved for antiproliferative effect, both of them are used in clinical practice for either symptom control or as antitumour agents in symptomatic or asymptomatic unresectable NETs. The value of somatostatin analogues therapy is unknown in the treatment of non-functioning pancreatic tumours and a few studies have been published in this field. Recently, a case report from a patient with metastatic non-functioning pancreatic tumour that was treated with octreotide-LAR showed that the treatment reduced the growth and progression of the tumour.
In a case report, octreotide LAR treatment for 9 months in the patient with multiple gastric carcinoids, led to the normalization of serum gastrin levels and the disappearance of tumours. In another study, five patients with gastric carcinoid and hypergastrinemia were treated with injection of octreotide LAR monthly for one year. Although, with this treatment the level of gastrin was not completely normal, but a significant reduction in tumour size and normalization of chromogranin A levels occur. We report a young patient with Multiple Endocrine Neoplasia (MEN) Type 1 with multiple gastric and pancreatic neuroendocrine tumours that was treated with monthly injection of Sandostatin LAR before and After Distal Pancreatectomy and partial gastrectomy and now it is well after four years of treatment with Sandostatin LAR.
Reference:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9672213/


